Incorporating RNA-Seq into AUGUSTUS
This document describes a method for
structurally annotating a genome based on deep sequencing of a transcriptome, RNA-Seq.
The guide below is a description of the method we initially
developed for and applied in the RNA-Seq based Genome Annotation
Assessment Project (rGASP).
It was applied to a variety of data types, such as from Illumina and
SOLiD, for read lengths between 33bp and 100bp, single and paired-end
reads, stranded and unstranded reads, on human, Drosophila and
C.elegans. We therefore expect this to be generally applicable to
other projects with some parameter adjustments. The general approach
is to generate hints for Augustus from the RNA-Seq, which can be used together
with hints from other sources if available (like from an existing
gene models, ESTs, protein or genomic conservation, MS/MS).
You may adjust below procedure for your needs, for example, if you choose to first assemble RNA-Seq
reads to contigs, you may just just treat them as other larger transcript fragments such as ESTs and use
BLAT/GMAP+blat2hints.pl to generate the hints.
1. Align RNA-Seq Reads to Genome (directly)
For species with many multi-exon genes
(most eukaryotes), we found that spliced reads are by far most
valuable. Spliced reads are reads that span an exon-exon boundary
and therefore delineate a complete intron. It is therefore important
to chose an alignment method that recovers as many true spliced reads
as possible.
Illumina:
In lack of a better program, we chose
to use the program BLAT, which is not customized for NGS reads (Next
Generation Sequencing), but found more introns than Tophat 1.0.11. However,
it is straightforward to adjust this pipeline to Bowtie/Tophat input,
because Tophat produces results in the same or similar format (wig
file and intron list including multiplicities).
blat -noHead -stepSize=5 -minIdentity=93 genome.masked.fa rnaseq.fa ali.psl
Then filter alignments for alignment
quality, uniqueness and pairedness, if applicable.
cat ali.psl | filterPSL.pl --uniq --paired > ali.f.psl
This filter requires by default a
minimum coverage of 80% of the read length and a minimum percent
identity of 92%.
--uniq
makes the filter script keep only alignments, which are 1) the best
alignment for that read and 2) unique in the sense that all other
alignments of that read have a score that is at most 0.96
(uniquethresh) the score of the best. The rationale behind stringent
filtering is that self-similarity of the genome or of transcripts in
particular may otherwise yield many false positive alignments. False
positive alignments can be a greater problem to Augustus as false
negative alignments, the later may frequently be corrected by the ab
initio model aided by neighboring alignments.
--paired
is another filter that only applies to paired-end reads. It requires
that two reads from the same read-pair (when the read names end in
.f,.r or /1,/2) are on opposite strands, on the same genomic contig
and at most maxintronlen (default: 500000) apart from each other.
For single-end reads just run
cat ali.psl | filterPSL.pl --uniq > ali.f.psl
Repeatmasking alone usually does not filter out
repetitions of DNA to the extend that RNA-Seq read mapping become
unambiguous or false positive. From our experience it can happen that even with the stringent
--unique filtering reads map to a likely wrong place in the genome. This is
frequently a region that is not a gene but shows high genomic similarity to
another region with a gene. You may therefore consider increasing the
stringency of the filtering by removing hits from regions that are repeated
similarly in the genome (and are not repeatmasked nonetheless). We chose to
remove hints that overlap a self-chain alignment of the UCSC Genome Browser.
Sort alignments by target sequence and
within target sequence by position for further processing (fs = filtered and sorted)
cat ali.f.psl | sort -n -k 16,16 | sort -s -k 14,14 > ali.fs.psl
[+]
SOLiD...
To align color-space reads we chose SHRIMP, e.g. in the case of 36bp reads we ran
rmapper-cs -s 111110011111 -n 2 -t 4 -o 5 -r 36 rnaseq.cfasta genome.masked.fa > shrimp.out
sorted the output
cat shrimp.out | grep -P "^>" | perl -ne 'split /\t/; print if ($_[1] !~ /_/)' | sort -k 1,1 > shrimp.s.out
and then filtered with
cat shrimp.s.out | filterShrimp.pl --minScore=320 --uniq --uniqthresh=0.9 > shrimp.f.out
Finally, also sort by target sequence
(chromosome):
sort -k 2,2 shrimp.f.out > shrimp.fs.out
[-]
Iterate Spliced Alignment (optional but recommended)
Individually aligning RNA-Seq
spliced reads to a genome is a hard problem and often
not unambiguously possible. Reads that span an intron will often not be aligned correctly,
in particular if only a short part of a read is in one exon and the larger part in another exon.
We use the knowledge on introns from the spliced alignment of other reads as well as from an
initial gene prediction to align spliced reads that are otherwise not aligned correctly.
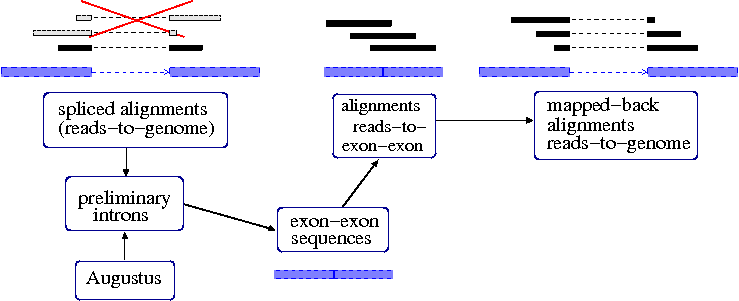
Procedure for iteratively finding spliced reads.
We compile a
preliminary set of introns. One part of this set is the
introns from above spliced alignments.
blat2hints.pl --intronsonly --in=ali.fs.psl --out=hints.introns.gff
Another (optional) part of the set is the set of introns predicted by ab initio
Augustus (or any other available structural genome annotation).
augustus --species=human --introns=on genome.masked.fa | tee \
aug.prelim.gff | grep -P "\tintron\t" > aug.introns.gff
We now join all unique preliminary introns in a list of the format
chr10:8749-10821:
cat hints.introns.gff aug.introns.gff | perl -ne 'split; print "@_[0]:@_[3]-@_[4]\n";' | sort -u > introns.lst
Next, we create a file
exex.fa with
exon-exon sequences. For each intron, we concatenate pieces of the flanking exons.
intron2exex.pl --introns=introns.lst --seq=genome.masked.fa --exex=exex.fa --map=map.psl
The resulting file
exex.fa looks like this:
>exex246182 75-GTAG-75 chr3.:100000692-100002450
GTTTGGCATCATGGGATGGTATTTTAGACTTGCCAGAACAGAACACTATTCACAAAGATTGCCTGCAGTTTATTGACCAGCTTTCAGTGCCAGAGGAGAAGGCAGCAGAATTACTTTTGGATATTGAATCTGTAATTACCTTTTATTGTA
The file
map.psl contains the positions of the exon-exon sequences in the genome in order to
back-map alignments of reads to
exex.fa that we will make next to alignments to the genome.
We align the original set of reads to the exon-exon sequences.
blat -noHead -stepSize=5 -minIdentity=93 -minScore=60 exex.fa rnaseq.fa exex.blat.psl
Now, we create read-to-genome aligments from the read-to-exon aligments that actually cross
the intron border:
pslMap.pl --filterunspliced --minmatch=60 --in=exex.blat.psl --map=map.psl --out=spliced.psl
Each read has been aligned twice now, to the genome and to the exon-exon sequences. From the
read-to-genome alignments we only take the
unspliced ones (seventh column < 35), as the spliced ones are
represented in
spliced.psl.
cat ali.psl | perl -ne 'split; print if ($_[7] <= 35);' | \
cat /dev/stdin spliced.psl | sort -k 10,10 | filterPSL.pl --uniq --paired > remap.f.psl
Finally, we sort the alignments by target sequence names for hint generation:
cat remap.f.psl | sort -n -k 16,16 | sort -s -k 14,14 > remap.fs.psl
As the code in this box is optional we rename the new alignments to
ali.psl
so people who did not read this box are not affected.
mv ali.fs.psl ali.direct.fs.psl
mv remap.fs.psl ali.fs.psl
2. Hint Generation
We currently generate two types of hints from the RNA-Seq alignments, exonpart and intron hints. Intron hints are more important but can currently not be generated by all NGS mapping programs: Most of them perform no spliced alignment.
Intron hints
blat2hints.pl --intronsonly --in=ali.fs.psl --out=hints.introns.gff
If you chose a different approach,
produce an output format like this for the intron hints:
chr1 b2h intron
14800 14877 0 . . mult=47;src=E
In this tab-separated format the
14800th base of sequence chr1 is
the first base of the intron, 14877 is the last, 47 is the
multiplicity of the intron and E is the source identifier for
setting the bonus of this evidence source below. The strand of an
intron will be determined automatically by Augustus.
Exonpart hints and wiggle
track
Each exonpart hint specifies an
interval that is likely to be exonic. We take the local coverage
depth into account. For the purpose of exonpart hint generation and
visualization we first summarize the alignments to a coverage depth
function in wiggle format. Wiggle format (.wig) is a format of the
UCSC Genome Browser.
aln2wig -f ali.fs.psl > cov.wig
This command works both for Illumina
and SOLiD reads. For SOLiD you replace ali.fs.psl
with shrimp.fs.out. If you
have stranded reads, you should generate two wig tracks, e.g.
plus.wig and minus.wig
and treat them separately below. The corresponding hints
should then have the strand column set. This will help Augustus to
determine the strand of a gene fragment.
Now generate the exonpart hints with
cat cov.wig | wig2hints.pl --width=10 --margin=10 --minthresh=2 --minscore=4 --prune=0.1 --src=W --type=ep \
--UCSC=unstranded.track --radius=4.5 --pri=4 --strand="." > hints.ep.gff
or adjust this command line accordingly
when you have a stranded wig file. This command generates for each
coverage island a sequence of adjacent 20bp exonpart hints
(exonpart = ep) and also stores the average coverage depth in each
such window as a means to adjust the weight of the evidence later.
Joined hints
Concatenate all hints:
cat hints.introns.gff hints.ep.gff [hints.other.gff] > hints.gff
The
optional hints.other.gff may contain hints from other evidence
sources. It may be preferable for efficiency to split the hints file
by chromosome.
3.
Run Augustus
3.1 Set hint parameters
Adjust the file extrinsic.cfg that
holds the hint parameters. Start by copying the file
config/extrinsic/extrinsic.M.RM.E.W.cfg. In the example above the
relevant, non-neutral lines could look like this:
[SOURCES]
M RM E W
[GENERAL]
ass 1 1 0.1 M 1 1e+100 RM 1 1 E 1 1 W 1 1
dss 1 1 0.1 M 1 1e+100 RM 1 1 E 1 1 W 1 1
exonpart 1 .992 .985 M 1 1e+100 RM 1 1 E 1 1 W 1 1.02
intron 1 .34 M 1 1e+100 RM 1 1 E 1 1e4 W 1 1
CDSpart 1 1 .985 M 1 1e+100 RM 1 1 E 1 1 W 1 1
UTRpart 1 1 .985 M 1 1e+100 RM 1 1 E 1 1 W 1 1
nonexonpart 1 1 M 1 1e+100 RM 1 1.15 E 1 1 W 1 1
The exonpart malus of .992 means
a weak penalty factor for every predicted exonic base that is not
supported by any exonpart hints. The exonpart bonus for hints of
source W of 1.02 means that gene structures get this bonus factor for
every exonpart hint of multiplicity 1 that is completely included in an
exon. Introns that are not supported by any intron hint are
penalized by .34, and introns that are supported by RNA-Seq hints are
rewarded by a factor of 10,000. The 0.985 are local
malus factors. The concept of a local malus was accounts for different levels of missing information, that becomes
more important with with higher general coverage of RNA-Seq. Like the (normal)
malus, the local malus also applies to exonic bases that are unsupported by
hints. In contract to the normal malus, the local
malus only applies to exons, that are well-supported at some region and not
supported at another region of the same exon. The following picture
illustrates a typical case where the local malus applies.
 This UTR exon candidate is very unevenly supported by RNA-Seq coverage and therefore likely to be too
long. For every base that is not covered by any exonpart hint a local UTRpart malus
factor of 0.985 is applied in addition to the normal exonpart malus
of 0.992.
This UTR exon candidate is very unevenly supported by RNA-Seq coverage and therefore likely to be too
long. For every base that is not covered by any exonpart hint a local UTRpart malus
factor of 0.985 is applied in addition to the normal exonpart malus
of 0.992.
The nonexonpart bonus of the RM source in this case is for
incorporating hints from repeat masking (src=RM).
Augustus is run on unmasked sequence with the repeat mask intervals given as nonexonpart
hints. In this regions, introns and intergenic regions are rewarded by
1.15 for each base. When run on softmasked genomes (in which the repeats are in lower case),
with the option --softmasking=1 then hints are generated automatically internally.
Above parameters have to be seen as an
example. The optimal values will depend on various parameters,
including
1) Amount of RNA-Seq and percentage of
genes expressed in the library. The more transcripts are expressed
and covered by RNA-Seq the stronger the malus can be. In the extreme
(hypothetical) case, all exonic bases are covered with RNA-Seq and
unsupported predicted exonic regions can be punished hard.
2) The stringency of filtering. If, for
some other reason, you have chosen to include more than one
hit of a read or you allow best alignments also when they are
ambiguous, then you may get a significant false positive rate of
hints and you may have to reduce bonus factors significantly.
3) Alignment method.
3.2 Run Augustus
augustus --species=human --UTR=on --extrinsicCfgFile=extrinsic.cfg --alternatives-from-evidence=true \
--hintsfile=hints.gff --allow_hinted_splicesites=atac genome.masked.fa
We recommend switching the UTR flag to
"on" because RNA-Seq covers UTR as well. With --UTR=off,
the exonpart hints would be (mis)interpreted to be hints for coding
parts of exons. --allow_hinted_splicesites=atac
allows Augustus to predict the (rare) introns that start with AT and
end with AC in addition to the GT-AG and GC-AG introns that are
allowed by default. --alternatives-from-evidence=true
turns on the prediction of alternative splicing. For this, intron
hints are in particular informative, as exonpart hints alone
will (if unstranded) not yield alternative transcripts.
Mario Stanke, 1/8/2012